Новое лекарственное средство, тестируемое как средство борьбы с раковыми заболеваниями,
способно помочь детям, страдающим прогерией Гартингса – болезнью, при которой происходит
стремительное преждевременное старение организма.
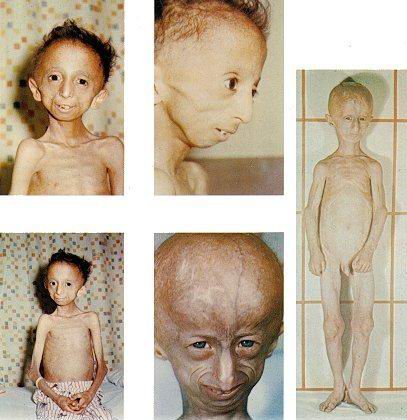
Дети, страдающие прогерией, выглядят нормальными в течение 6-12 месяцев после рождения.
После этого у них развиваются симптомы, характерные для преклонного возраста: морщинистая
кожа, облысение, ломкость костей и атеросклероз, что, как правило, приводит к их гибели в
возрасте около 13 лет. В настоящее время адекватного лечения этого заболевания не существует.
Однако результаты, полученные исследователями университета имени Джона Хопкинса,
США и подтвержденные рядом других лабораторий, вселяют надежду, что препараты группы
ингибиторов фарнезил трансферазы (FTI) способны предотвратить развитие процессов старения
в культурах клеток, аналогичных клеткам больных прогерией. Их ядра, в отличие от нормальных
ядер округлой формы, состоят из многочисленных долей и иногда даже имеют вид виноградных
гроздьев.
В лабораторных условиях применение FTI, уже проходящего клинические испытания на
онкологических пациентах, возвращало ядрам «стареющих» клеток нормальную форму. При этом
препарат блокирует синтез дефектного белка, вызывающего синдром, на ранних этапах его
формирования. Однако еще не известно, достаточно ли возвращения ядрам их нормального
внешнего вида для предотвращения или хотя бы замедления развития заболевания.
Исследователи занимаются изучением процессов преобразования и модифицирования белков
внутри клетки с целью разработки методов лечения онкологических заболеваний уже на
протяжении двух десятков лет. То, что в развитии прогерии участвует один из изучаемых ими
белков – ядерный протеин ламин А - оказалось своего рода сюрпризом.
Действие тестируемых препаратов FTI направлено на блокирование на ранних стадия
синтеза определенных белков в клетках дрожжей и млекопитающих. Изучением этого процесса
авторы исследования занимаются уже более 20 лет.
Процесс начинается с присоединения к полностью сформированной белковой молекуле
липидной группы, называемой фарнезил, недалеко от конца белковой цепочки. После этого к
соседней аминокислоте присоединяется крошечная метильная группа. И, наконец, некоторые
белки, модификация которых происходит описанным способом, проходят дополнительный этап.
При этом модифицированный концевой участок молекулы и прилегающие 15 аминокислот
отрезаются с помощью фермента (FTI), обнаруженного авторами.
У дрожжей полностью модифицированный таким образом протеин необходим для
нормального процесса размножения, причем важную роль при этом играет меньшая часть
молекулы, несущая на себе модификации. В клетках млекопитающих, напротив, образующийся
при процессинге ламина А больший отрезок белковой молекулы, не несущий на себе
модифицирующих групп, необходим для нормального функционирования и поддержания
структуры клеточного ядра.
У детей, страдающих прогерией, в результате мутации отсутствует участок молекулы ламина
А, включающего в себя критическую точку, по которой происходит отрезание
модифицированного участка. Хотя теоретически существует множество возможных вариантов
формирования патологического ламина А, именно сохранение модифицированного участка белка
является причиной развития характерных для заболевания аномалий.
Для проверки гипотезы были созданы генетически модифицированные клеточные линии.
Клетки одной линии несли мутацию, нарушающую процессинг ламина А в самом его начале – на
этапе присоединения фарнезила. В клетках второй линии был нарушен процесс расщепления
белковой молекулы на два отрезка.
Ни та, ни другая линии не содержали нормального ламина А, но проблемы, характерные для
клеток больных прогерией, были отмечены только в клетках, содержащих молекулы ламина А от
которых не был отщеплен участок, несущий фарнезил. В клетках, содержащих аномальный, но не
несущий фарнезил, ламин А, ядра имели совершенно нормальную форму.
Таким образом, была отвергнута устоявшаяся гипотеза, гласящая, что в основе заболевания
лежит дефектный ген, ответственный за синтез изначально аномального белка – прогерина.
При внесении в культуру дефектных клеток препарата FTI наблюдался ожидаемый эффект –
их ядра приобретали нормальный вид. Так как препарат проходит клинические испытания и
хорошо переносится онкологическими пациентами, можно рассчитывать на его скорое
включение в тестирование на возможность лечения больных прогерией.
Онкологические заболевания встречаются гораздо чаще, чем синдром прогерии, поражающий
в среднем одного из 8 000 000 новорожденных в США. Обычно такие редкие заболевания не
привлекают внимания разработчиков лекарственных препаратов, но счастливое совпадение, что
заболевание вызывается белком, для модификации которого необходим фарнезил, делает
возможным использование уже существующего препарата.
Источник новости: Bio.com